We are currently trying to generate a stability contour plot using pymatgen. The examples online seem to be using an older version of the code where class PourbaixAnalyzer was used. We are simply trying to get the stability of a compound based off its pH, concentration, and the applied voltage. We are trying to use the function:
PourbaixDiagramObject.get_decomposition_energy(PourbaixEntry,pH,V)
as a replacement to the function in the example:
PourbaixAnalyzerObject.get_entry_stability(Entry, pH,V)
We are getting energies around -1–10 eV/atom. On the provided example energies range from 0-1. Are we using the wrong function? Is there a correction to the decomp energy we need to account for?
Hi ZKaiser,
Thanks for bringing the outdated notebook to our attention. You can see an updated notebook here, which I’ll double-check later this evening. I’ll also look into why the pymatgen documentation one is out of date.
Do you mind sharing a snippet of code that’s giving you this issue? My guess is that either (a) the solid PD filter is causing a problem, which you can disable with filter_solids=False or (b) that there’s an issue with the entry itself, which I might be able to address if you can provide me with more detail.
Also, if you don’t mind, can you tell me how you arrived at the example notebook you linked? The current pymatgen website points towards the matgenb repo, but I suspect there are other avenues to the old examples we should probably take down since they’re deprecated.
Hi Joseph,
Thanks for the reply!
The code I am running is as follows:
#set up MPRester
mpr = MPRester(“MY_API_KEY_HERE”)
#Find entries of desired compound
entryP=mpr.get_entries(“mp-1692”)
#Find elements in compound
elementarrayobj=entryP[0].composition.elements
elementarray = []
#Make string array of elements
for z,h in enumerate(elementarrayobj):
elementarray.append(h.symbol)
#pass string of elemets to make pourbaix diagram
pbd = PourbaixDiagram(mpr.get_pourbaix_entries(elementarray))
#make the Pourbaix Plotter
pbp = PourbaixPlotter(pbd)
#print out entry id
print(entryP[0].entry_id)
#Generate pourbaix entry for specific compound
pbe = PourbaixEntry(entryP[0])
#print out decomp eneryg
print(pbd.get_decomposition_energy(pbe,pH=7,V=-0.2))
#plot
pbp.plot_entry_stability(pbe)
pH_range=[-2,16]
pH_resolution=100
V_range=[-3,3]
V_resolution=100
pH, V = np.mgrid[pH_range[0]:pH_range[1]:pH_resolution1j,V_range[0]:V_range[1]:V_resolution1j]
At pH=7 and V=-0.2 I am getting an energy of -8.91
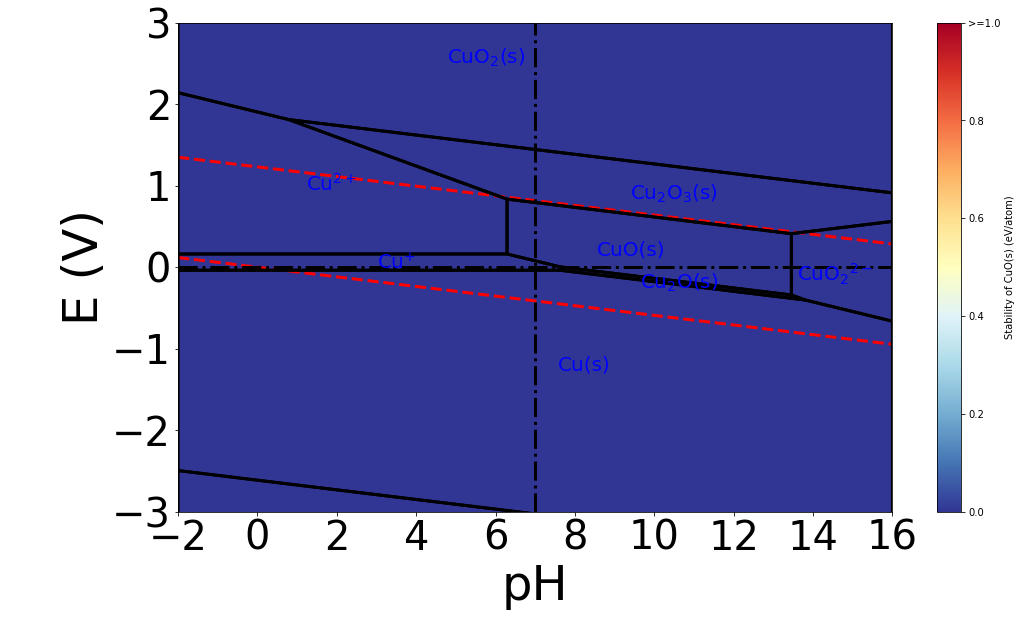
The Pourbaix diagram generated is shown below
When running your code it worked perfectly fine.
I think the problem may be in how I generate the PourbaixEntry. You do mpr.get_pourbaix_entries(chemsys) while I just use the constructor PourbaixEntry(entry), though it seems to me that both methods should work.
I got to the old notebook by simply Googling “Pymatgen Pourbaix”. It is the first result, though now it gives a 404 error.
MOD EDIT: Removed API Key from provided snippet, please do not post your API key.
1 Like
Thanks for the snippet. The way the pourbaix diagram code works is still a bit more convoluted than I’d like, the issue here is that valid PourbaixEntries require some preprocessing such that the input entry energy is the formation energy, rather than a raw unreferenced energy from VASP (or any other electronic structure code). This preprocessing is done in the MPRester.get_pourbaix_entries method but not the MPRester.get_entries method. If you’re using our data, I’d recommend getting the entry you’re looking for using the list comprehension in the example, e.g.
pourbaix_entries = mpr.get_pourbaix_entries(["Cu"])
entryP = [e for e in pourbaix_entries if e.entry_id=="mp-1692"][0]
pbd = PourbaixDiagram(pourbaix_entries)
#make the Pourbaix Plotter
pbp = PourbaixPlotter(pbd)
etc.
1 Like
Hi,
I was trying to calculate the aqueous decomposition stability for InP using the PourbaixDiagram routine in pymatgen. The script is as follows:
entries = (mpr.get_pourbaix_entries([“In”, “P”]))
species = [entry for entry in entries if entry.entry_id==“mp-20351”][0]
pbx = PourbaixDiagram(entries)
print pbx.get_decomposition_energy(species, pH=9, V=1.5)
The outcome I am getting is 10.1652846824
This does not match the number provided for InP in the paper Chem. Mater. 2017, 29, 10159-10167 by Singh et. al., which is around 8.01 (pH = 9 and V = 1.5)
Please let me know if I am missing something. Thank you very much.
Regards,
Naiwrit.