I downloaded a few structures from the database about a year and a half ago, and this week I re-downloaded the same structures again. I’ve noticed that several structures that used to have small band gaps are now metallic – same MPID, same phase, same energy above the hull.
One example is the material LiMgP, mp-10178. My old copy of the database reports a band gap of 217 meV; currently it says it is metallic. Can you confirm that this data has changed recently (and that it wasn’t just a bug on my end), and provide the justification for why? I am now worried that the electronic structure data is not rigorously calculated or trustworthy.
Hi there – a follow-up message on this. By my count, there are 1,842 lithium containing materials alone whose band gaps have changed in the last year or so, and in 747 of these cases the structures have become metallic. This seems like it must be a bug; I can’t imagine a structure like mp-767354, which has 14 oxygens per formula unit, would be metallic.
We keep calculating and adding more data to MP database. The band structure calculation of mp-10178 was finished on 2015-1-31, and had been added into the database thereafter. So that if you download the data earlier than that date, it was the band gap from the eigenvalues of static run.
The calculations for each material normally has four stages: structure optimization -> static run -> DOS calculation -> Band structure calculation, in this order.
Before completion of the four-stage workflow, we can already take a glance at some band properties of a material by analyzing its eigenvalues of static run, that said, at this point, the calculation result comes from a loose K-point mesh run instead of the dense line-mode mesh mode in real band structure calculation. Hence, we can obtain some info of energy bands properties, but compromise some of the accuracy at that stage.
When a full workflow finished, we will have the new band structure data available, and the band structure and DOS will be published on MP website. Therefore, the new band gap of 0eV comes from the line-mode k-point sampling and is more accurate and reliable. We still keep cranking the reel to have more structure calculated. Please stay tuned.
Compounds with oxygen can be conductive. E.g., Yttrium barium copper oxide (YBCO) is an oxide and superconductive; Indium tin oxide (ITO) is also conductive too.
Also keep in mind that all the energy bands on MP comes from GGA or GGA+U calculations. So that there is an under estimation there. It may not able to capture some band gap opening from Spinel orbital interaction as well.
Thanks for the prompt response. This makes sense – I can see how the gap value would change after calculating the full DOS + denser k-mesh. However, I always thought that calculating the gap with these more rigorous methods will always either result in an equal or lesser value of the gap than the static run calculation would give you, since it will expose more features of the band structure than the static run (maybe this is not a correct assumption?). My count shows 540 Li containing materials have had their gaps increase since 2015, and 37 of them by more than 1 eV. Please forgive my ignorance, as I’m not an active DFT user, but is this sort of change to be expected?
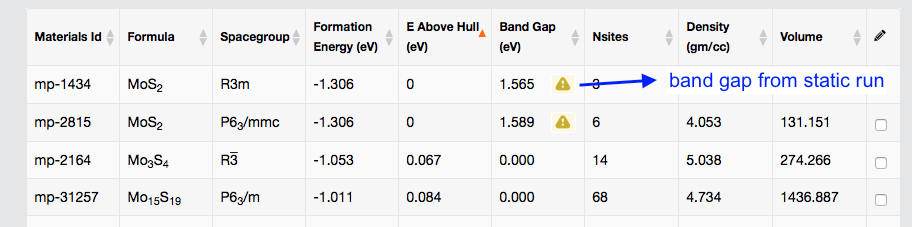
Yes, this type of change will keep happening in the future as more and more bands calculations is underway. The indication of the band gap accuracy can be found from MP site too.
In above searching table, the exclamatory mark indicates it is less accurate. The lower two have the better data quality from denser k mesh run.
Also in each material detail page, band structure and DOS plots also indicate the completion of the band structure run. If they do not show up, the calculations are not finished.