hello,
when I try to get vasp inputs(INCAR, POSCAR, POTCAR, KPOINTS) by ‘from pymatgen.io.vasp.sets import MPRelaxSet’ in pymatgen . I got some error(OSError: You do not have the right POTCAR with functional PBE and label Nd_3 in your VASP_PSP_DIR), my VASP_PSP_DIR is /home/henkekao/POT-VASP/PAW-2003/pot-pbe/.
Can you show me a example?
What is the right POTCAR with functional PBE?
Should I rename the pseudo potential files to specific name?
A reply would be highly appreciated.
Hi Henkekao,
I think you should be able to set your PSP directory using the following command.
pmg config --add PMG_VASP_PSP_DIR /home/henkekao/POT-VASP/PAW-2003/pot-pbe/
If this doesn’t work, try using the more detailed instructions in pymatgen’s documentation. Let us know if you encounter any further issues.
1 Like
Thanks
I figure out the reason! When I altered the /home/henkekao/POT-VASP/PAW-2003/pot-pbe/ to /home/henkekao/POT-VASP/PAW-2003/POT_GGA_PAW_PBE. And then
pmg config -p /home/henkekao/POT-VASP/PAW-2003/POT_GGA_PAW_PBE /home/henkekao/potcar/POTCAR
pmg config --add PMG_VASP_PSP_DIR /home/henkekao/potcar/POTCAR
The error is gone!
So, I think a specific directory name is needed.
1 Like
Hi @Joseph_Montoya
I find some strange in pymatgen.io.vasp.sets.MPRelaxSet.
For example:
from pymatgen.ext.matproj import MPRester
from pymatgen.io.vasp.sets import MPRelaxSet
MP = MPRester(‘XXXXXXXXXXXXX’)
a = MP.get_structure_by_material_id(‘mp-19306’)
b = MPRelaxSet(a)
b.write_input(’.’)
But, when I compare the files(INCAR, KPOINTS,POSCAR, POTCAR) got in those commands with what you give on the website, I find they are different. Especially some parameters in INCAR (MAGMOM) and KPOINTS.
Hope for your reply.

Hi @henkekao,
The MAGMOMs are changing because the structure that’s being returned by MP.get_structure_by_material_id is the final structure from our optimization calculation, which includes the MAGMOMs in the result of that calculation a site property. The MPRelaxSet will take the magmoms attached to that structure and use them in the writing of the vasp files. It shouldn’t make a significant difference in the final result of a given calculation, but if you’d prefer to use a clean structure to start, you can do either of the following:
First, if you’d prefer to use the initial structure from the calculation, you can use
a = MP.get_structure_by_material_id('mp-19306', final=False)
and the initial structure that was used in the calculation (without the MAGMOMs) attached will be returned.
If you’d prefer to use the final structure without the MAGMOMs from our calculation attached, you can unset the site_property of the MAGMOMs using the following:
a = MP.get_structure_by_material_id('mp-19306')
a.remove_site_property("magmom")
I’m not seeing a difference in the KPOINTS file or EDIFF parameters on my machine (relative to this set of calculation details), can you specify which calculation details page you’re looking at where you’re seeing a discrepancy?
Hi @Joseph_Montoya
Thanks for your reply!
I think I have known where I’m wrong. I mistake final structure as the initial structure. So, I will get a different KPOINTS and EDIFF.
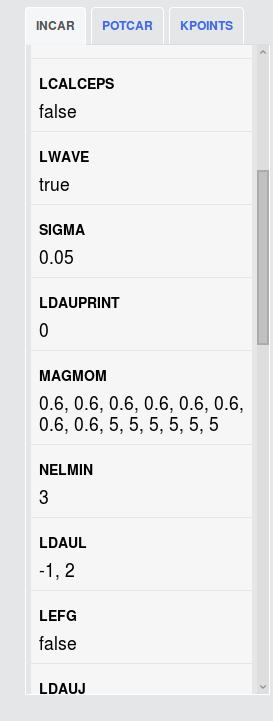
One more question. Among those calculations ,which one is the initial structure? And what is the difference among them?
Thanks
Best
Tao Zhang
The initial/final structures should be yielded in the bolded GGA+U optimize structure (2x) (the one in the top row). The remainder of the tasks (static, uniform, etc.) use different input sets (specifically, MPStaticSet and MPNSCFSet) and should only be using the final structure as input and output.
Hi @Joseph_Montoya:
Sorry to bother you again.
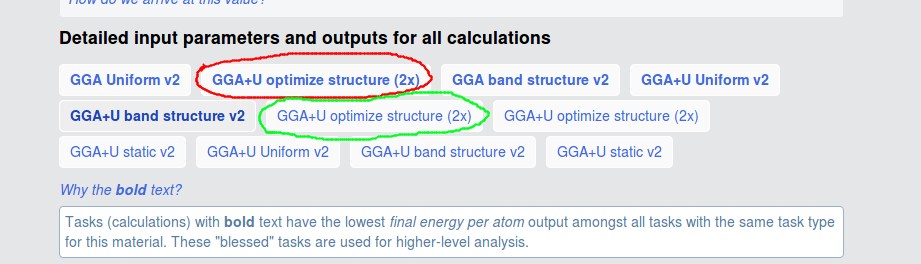
I am still confused about all the calculations. For example, what is the difference between the two calculations mark with red circle and green circle? I find the MAGMOM in INCAR and their initial structure are different. And the total energy of the two calculations are different, red circle is -93.935 eV, green circle is -92.944 eV. What caused this deviation?
I also calculate both structures with the pymatgen recommend vasp set. But the pymatgen recommend MAGMOM for red circle is MAGMOM = 6*5, 8*0.6, differ from shown on the website 5, 5, 5, -5, -5, -5, 0.6, 0.6, 0.6, 0.6, 0.6, 0.6, 0.6, 0.6 . So, I calculate the red circle structure in two kinds of sets. The is results shown below:
red circle:
- MAGMOM = 6*5, 8*0.6 total energy is -93.085 eV
- MAGMOM = 5, 5, 5, -5, -5, -5, 0.6, 0.6, 0.6, 0.6, 0.6, 0.6, 0.6, 0.6 total energy is -93.581 eV
green circle:
total energy is -93.047 eV
So, even if I used the same setup, I still couldn’t get the same result. Especially for the red circle, because the red circle is chosen as the lowest total energy.
Hope for your reply.
Best
Tao Zhang
The green and the red circles have different initial structures (at least sufficiently different to not be recognized as equivalent by our algorithms), but relaxed to the same structure, so are assigned to the same material. The text in your picture explains that we “bless” certain tasks (i. e. the ones that yield lower energies) and those are highlighted in bold (i. e. the red circle).
The recommended VASP input set doesn’t include considerations of magnetic orderings and occasionally materials on the website will have multiple magnetic orderings that have been tested to determine which one is the ground state. The materials details page specifies that the ground state magnetic ordering is ferrimagnetic (not ferromagnetic or antiferromagnetic, which I believe the spin states you’ve indicated in your post correspond to), and the final structure that the MPRester delivers shows that the final magnetic moments are:
[3.921, 4.009, 3.924, 4.242, -4.195, -4.195, 0.035, 0.055, 0.01, 0.055, 0.037, 0.01, 0.035, 0.037]
Which probably means we started with 5, 5, 5, 5, -5, -5, 0.6, 0.6… etc.
Hi @Joseph_Montoya:
Thanks for your reply again.
Today, I suddenly remembered this question and I try to calculate the initial structure with MAGMOM = 5, 5, 5, 5, -5, -5, 0.6, 0.6……, then the total energy is -93.94607058 eV. I think the deviation can be neglected.
The results indicate, the INCAR displayed on the web page may be wrong?
And, pymatgen.io.vasp.sets can only give a general INCAR, it will not contain magnetic ordering or some other specific properties?
Hope for your reply
Best
Tao Zhang
Hi @henkekao,
I believe the INCAR here is correct, it looks like the initial phase is AFM, but one of the -5 spins flips over the course of the VASP optimization procedure to produce the ferrimagnetic phase. Is that what you’re seeing in your calculation?
The VASP input set will give a general (i. e. FM) INCAR if the magmoms are not set in the input structure site properties. If those are set (as they are if you get the “final” structure from the MPRester), then the VASP input should reflect the structure magmoms.
Got the same problem, and solve with this solution
Hey,
Which programming language is this:
pmg config --add PMG_VASP_PSP_DIR /home/henkekao/POT-VASP/PAW-2003/pot-pbe/
I used it in python but it had syntax error.
@kamran_haddadian use the pmg command in your terminal (bash, zsh, …), i.e. it’s a command line program.
Thread closed due to inactivity, please open a new thread to address related issues.