I was working with the fix ttm/mod function. In the documentation of fix ttm on the website, the vacuum region in the fix ttm/mod is defined using the l_surface and r_surface parameters along the X direction (fix ttm command — LAMMPS documentation). I was inspecting the fix_ttm_mod.cpp file and this line in the below image confuses me. The force applies to all the atoms in the region of interest. But the conditions seem like, they are applied to the vacuum region. Can anyone clarify this? @akohlmey @sjplimp
Thank you
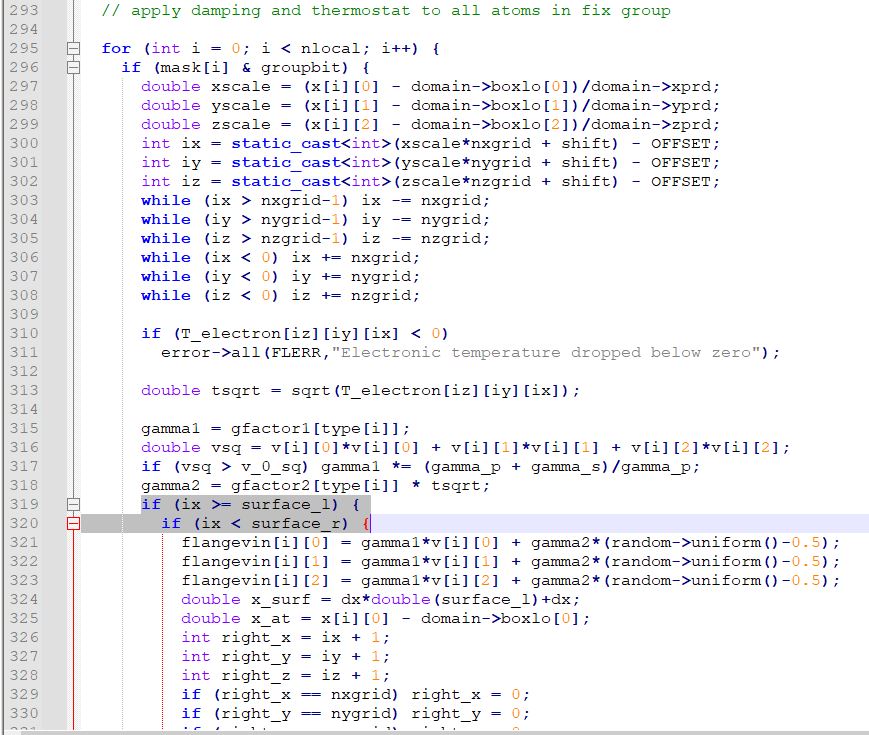
I think those variables correspond to the surfaces of the atomic system. That is, ttm cells between surface_l and surface_r have atoms/electrons and transport is evaluated.
1 Like
Hi @baerb , thank you for replying to the thread. It explicitly is stated in the documentation " The initial borders of vacuum can be set in the init_file via lsurface and rsurface parameters". This is confusing with the code implementation.
There can only be either atoms or vacuum. Doesn’t defining the bounds of one necessarily define the bounds of the other?
Edit: I suppose a better question is: do you think the ttm is doing something wrong or is your issue purely with the documentation?
Let’s say for example. The whole X grid is divided into 20 mesh points and I define 0 to 14 to be the atomic region and 15 to 19 to be the vacuum. If the l_surface and r_surface define the boundaries of the vacuum, the lsurface=15 and rsurface=19. If it is the other way around, then the lsurface=0 and rsurface=14. This is what I think. Correct me if I am wrong.
I agree that the documentation around defining the vacuum is a bit ambiguous, but reading the source code clears that up. This has to be the way to do it, otherwise you are performing operations on the vacuum region, as you said.
I think in an ideal world this feature would have been included in the example script, but I seem to recall it being a periodic solid.
1 Like
Thank you for the clarification. And yes, the example script utilizing the ttm/mod uses a periodic solid
How do you come to this conclusion?
What I see when I look at this code is that it applies a force to atoms between the left and the right “surface” of the electron grid.
You are discarding that fix ttm/mod requires a periodic system. Thus each “surface” has to be a vacuum/electron interface. Since the natural setup of such a system is to have the atoms in the center,
this would specify one interface to the left side of the box, i.e. vacuum/electron and one interface to the right, i.e. electron/vacuum.
Please keep in mind that LAMMPS “lives” off the contributions from many users in many countries and few of them are native speakers of english and sometimes subtle details like this can get lost in translation when the documentation is written by contributors whose programming skills are far better than their english language skills.
yes.
this would be the logical (to me) input.
If you think you can clarify this in the documentation, feel free to submit a pull request (and a tested simple demo example).
@akohlmey Thank you for replying to me. I was checking the output from ttm and ttm/mod using the same set of input parameters. When outputting the electron energy and transferred energy from the ttm and ttm/mod, they were very different. If I specified lsurface and rsurface region in the ttm/mod to be the vacuum region, the energy transferred is very small even with a very high value of electron-phonon coupling. And the output electron temperature remains almost the same with the initial input electron temperature. But this doesn’t happen when fix ttm is used. Therefore, I checked both the source codes of fix ttm and fix ttm/mod. The langevin thermostat is applied in the whole atomic region for the ttm case while for the ttm/mod case, it is the region defined between the lsurface and the rsurface region. So, it seems that the documentation is wrong about the lsurface and rsurface to the vacuum region. The correction would be: lsurface and rsurface to the atomic region or the source code has to be modifed so that lsurface and rsurface to the vacuum region.
I will submit a pull request in github today. Thank you!
That is just as wrong or correct as the current description.
Please keep in mind that you are using a very unusual setup where you have your atomic system exactly on the periodic box boundary and this makes one of the interface locations ambiguous. The more common setup would be to have vacuum above and below the slab with atoms and then the left / right surface requirement (a better discription would probably be “lower / upper interface”) is unique.
The current setup I am using for the simulation has an atomic system with a vacuum on one side along the (+)X direction and a periodic boundary condition in each direction of the bounding box. The current description of the ttm/mod allows me to have vacuum only along the X direction. I haven’t done any simulations with vacuums along both side of the atomic slabs.
You have to have periodic boundary conditions, thus your choice of origin is arbitrary.
There is no such thing with vacuum on “both sides”. There is still just one vacuum region.
As I already stated, in your specific setup the one interface location is ambiguous, since it is exactly on the box boundary.
My current script box is as below:
#------------------------ VARIABLES-----------------------------------
variable boxLx equal 50
variable boxLy equal 29
variable boxLz equal 32
variable latparam equal 3.180000
variable myT equal 300.000000
variable run1 equal 1000
variable run2 equal 5000
# ------------------------ INITIALIZATION ----------------------------
echo both
units metal
dimension 3
boundary p p p
atom_style atomic
atom_modify map array
# ----------------------- ATOM DEFINITION ----------------------------
lattice custom ${latparam} &
origin 0.1 0.1 0.1 &
a1 0.500000000000000 -0.866025403784439 0.000000000000000 &
a2 0.500000000000000 0.866025403784439 0.000000000000000 &
a3 0.000000000000000 0.000000000000000 1.63 &
basis 0.333333333333333 0.666666666666667 0.0 &
basis 0.666666666666667 0.333333333333333 0.5 &
basis 0.333333333333333 0.666666666666667 0.375 &
basis 0.666666666666667 0.333333333333333 0.875
region box block -${boxLx} ${boxLx} -${boxLy} ${boxLy} -${boxLz} ${boxLz}
create_box 2 box
create_atoms 1 box &
basis 3 2 &
basis 4 2
mass 1 69.723
mass 2 14.007
# ------------------------ FORCE FIELDS ------------------------------
pair_style hybrid/overlay tersoff zbl 0.6 0.8
pair_coeff * * tersoff GaN.tersoff Ga N
pair_coeff 1 1 zbl 31 31 #Ga-Ga
pair_coeff 2 2 zbl 7 7 #N-N
pair_coeff 1 2 zbl 31 7 #Ga-N
neigh_modify every 1 delay 0 check yes
thermo 500
thermo_style custom step dt time press pe temp lx ly lz
variable Potential equal "pe"
compute PE all pe/atom
compute KE all ke/atom
# ----------- relax system --------------------#
variable randomSeed equal 2749
timestep 0.001
velocity all create ${myT} ${randomSeed} mom yes
fix 1 all npt temp ${myT} ${myT} 0.1 iso 0.0 0.0 1 drag 1
run ${run1}
unfix 1
# ----------- add empty region along X --------------------#
change_box all x delta 0.0 150.0 units box
fix 1 all nvt temp ${myT} ${myT} 0.1
run ${run1}
unfix 1
reset_timestep 0
fix 1 all nve
fix 2 all dt/reset 1 1.0e-4 1.0e-3 0.05 units box
fix twotemp all ttm/mod 36521 GaN.ttm_mod 20 15 15 set 22000 outfile 1000 Temperature.out
thermo 1000
thermo_style custom step dt time temp f_twotemp[1] f_twotemp[2]
run ${run2}
unfix 1
unfix 2
unfix twotemp
I am attaching my input scripts with the output as well. If the lsurface and rsurface are set to be the region of the vacuum (lsurface =15, rsurface=20), the temperature in the electron grids remains almost constant with the initial set electron temperature. If the lsurface and rsurface are set to the atomic region (lsurface =0, rsurface=14), the electron temperature decreases with time which makes more sense to me. Please let me know if I am making any mistakes here.
ttm:mod.zip (203.8 KB)
I have nothing to add to my previous posts. We are going in circles now. The problem you have is a problem of your understanding of how the authors of fix ttm/mod assume people will set up their systems.
I have already pointed out that your choice of system geometry make the location of the interfaces ambiguous and this is why you seem to be misunderstanding things (or you don’t fully understand periodic boundaries or a combination of those). I cannot read minds, so I can only speculate about why you are insisting on your view and cannot make an effort to understand what I have been trying to explain. As I had pointed out, if you move your atoms into the center of the box, there is no more ambiguity and only one way to specify the location of the two surfaces/interfaces.
1 Like
Got it. Thank you! Let me try to run it that way.
Hi @akohlmey,
I ran as per your instruction for the periodic boundary. You can see the log files in the attached zip file. This is taken from the example of the ttm/mod provided in the example folder. The atoms have been moved to the center of the box and there should be no more ambiguity. I did two simulation one specifying the region between lsurface and rsurface to be the atomic region (logfile1) while the other one specifies the region between lsurface and rsurface to be vacuum (logfile2). If you see the first logfile1, you can clearly see the net energy transfer value over time. However, for the 2nd case, there is no net energy transfer value from the electronic subsystem to the lattice subsystem as there are no atoms. My point therefore is: there should be a change in the documentation regarding this, specifying what exactly the region between lsurface and rsurface contains. Otherwise, it is confusing what the description mentions about the lsurface and the rsurface.
ttm_mod.zip (18.0 KB)
The Si.ttm_mod2 input is incorrect. The region between index 21 and index 30 is covering only half the vacuum. The boundaries of the vacuum in your logic (and taking into account the periodicity) would be 21 for the left and 10 for the right, but LAMMPS will refuse that.
As I already pointed out, the whole problem appears to be a matter of translation to and from English. Neither you nor the authors of the fix are native speakers and thus have different connotations when looking at the documentation. You are (for a reason I don’t understand) fixated that the two interfaces have to be specified as the “boundaries of the vacuum”. But it is really just that you specify the position of the vacuum/atoms and the atoms/vacuum interfaces. And that can only be fulfilled without LAMMPS refusing it when you use the Si.ttm_mod1 input.
We have added a small rewording of the docs in the development branch that may reduce the risk of a misinterpretation which will be in the next release: fix ttm command — LAMMPS documentation
But the main step for you is that you realize your mistake and misinterpretation and accept how this was meant to be read which to me (as another non-native speaker) appears obvious. But that may just be a sign of my experience of communicating in English with researchers from many regions and countries of the world for a long time and thus taking into account that one has to look for the intention of the author of some comments or documentation instead of getting hooked on the semantics of a somewhat ambiguous wording.
1 Like